
华东师范大学/华东理工大学药学院的李诗良/李洪林教授团队在国际知名学术期刊Oncogene上发表了题为"Clustering single-cell RNA sequencing data via iterative smoothing and self-supervised discriminative embedding"的研究论文。这项研究提出了一种创新的深度学习聚类方法——scRISE模型,为单细胞RNA测序 (scRNA-seq) 数据的聚类分析提供了高效的解决方案。scRISE模型是一种巧妙地融合了迭代平滑技术和自监督判别嵌入技术的深度学习框架。该模型的架构由两个主要模块构成:迭代平滑模块和自监督判别嵌入模块,以协同优化单细胞数据的聚类分析过程。
单细胞RNA测序 (scRNA-seq) 技术是现代生物学研究的一大突破,它允许研究人员在单个细胞层面上测量基因表达,为我们理解细胞多样性和复杂生物系统的细胞组成提供了前所未有的视角。这项技术通过分析成千上万个细胞中成千上万个基因的表达模式,揭示了细胞间的变异性,有助于识别稀有细胞群体和推断细胞谱系关系。然而,scRNA-seq数据的高维度特性、技术噪声、数据缺失和批次效应等问题,为数据分析带来了一系列挑战。在scRNA-seq数据分析中,聚类分析是识别细胞类型和亚群的一个关键步骤,这要求我们要有精确的相似性度量方法和有效的算法来应对这些数据的复杂性。尽管目前已有多种聚类方法被提出,但在选择一个既稳健又能够准确反映细胞相似性的度量标准,以及从复杂的数据结构中恢复出真实的细胞类型方面,我们仍面临着一个尚未解决的难题。这一挑战要求我们不断地探索和改进算法,以期达到更高的数据分析精度和可靠性。为了提高聚类分析的准确性,研究团队开发了一种名为scRISE的新型深度学习聚类方法。scRISE模型的创新之处在于两个主要方面:
1、迭代平滑技术:scRISE采用了拉普拉斯平滑技术,通过迭代过程逐步优化数据,有效减少了高频噪声,提高了数据质量。
2、自监督学习:模型中集成的自监督判别嵌入模块,能够自动选择正负样本对,减少了人为干预,提高了聚类的准确性和效率。
通过这些创新技术,scRISE模型能够有效应对scRNA-seq数据的高维度和噪声挑战,从而提供更为精确和可靠的聚类分析结果。这不仅推动了单细胞数据分析技术的发展,也为生物学领域的研究者们提供了一个强大的工具,以探索和理解复杂的细胞生态系统,揭示细胞间的相互作用和通讯机制。
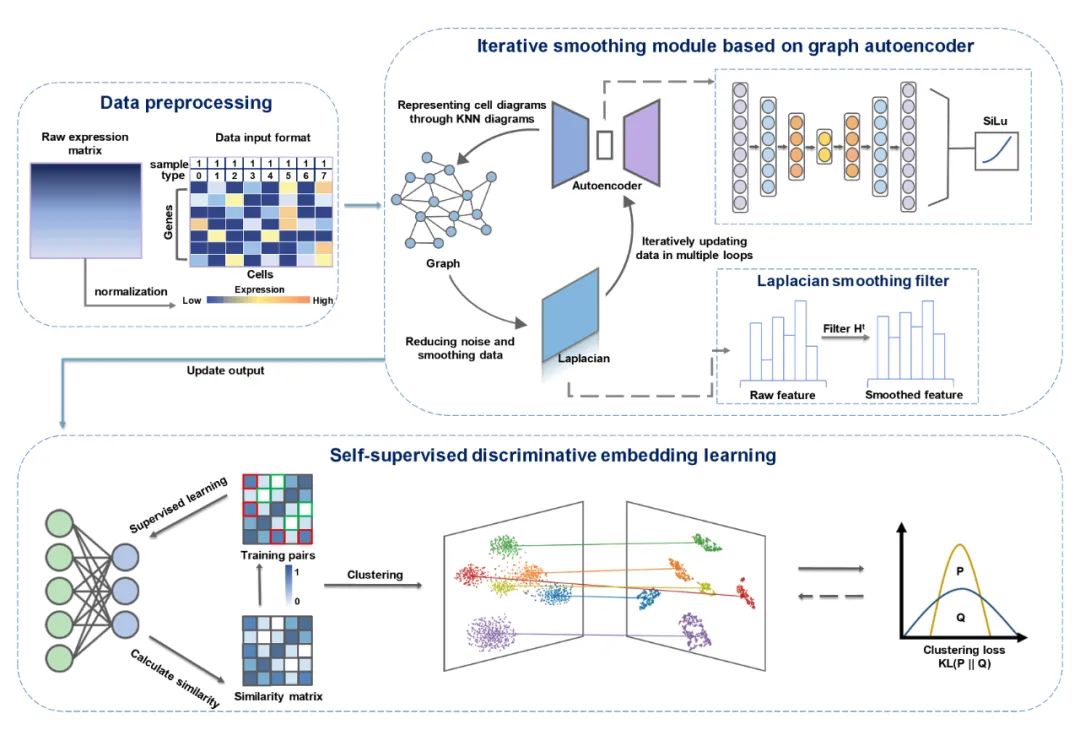
scRISE方法由两个主要模块构成,如图1所示。首先,研究团队使用了一个基于图自编码器的迭代平滑模块,该模块结合了自编码器和拉普拉斯平滑滤波器来平滑和重建可能存在噪声、不完整或粗糙的数据,同时融入了细胞间的结构信息。自编码器能够准确地重建数据中的主要信号,而拉普拉斯平滑滤波器通过平滑处理进一步改善数据质量,减少噪声的影响。这个迭代过程不断地更新重建数据,逐步提高单细胞数据的准确性和稳定性。接着,采用了一个自监督判别嵌入模块,该模块利用细胞间的相似性来选择正负样本对,以确定数据中固有的簇。在这个模块中,正负样本对的阈值是自适应选择的,这样属于同一簇的样本自然会被推到一起,而来自不同簇的样本则在嵌入空间中相互排斥。该模块旨在通过学习数据分布中固有的相似性结构来增强聚类性能。通过这两个模块的融合,scRISE有效地去除了数据中的不兼容和噪声信号,并实现了无需大量人工干预的自监督聚类。
为了评估scRISE方法相较于现有技术的优势,该论文将其应用于17个公开真实的scRNA-seq数据集上,与现有的聚类方法通过一些性能指标进行模型比较,这些指标包括调整兰德指数 (ARI) 和标准化互信息 (NMI) 。
1. 模型评价
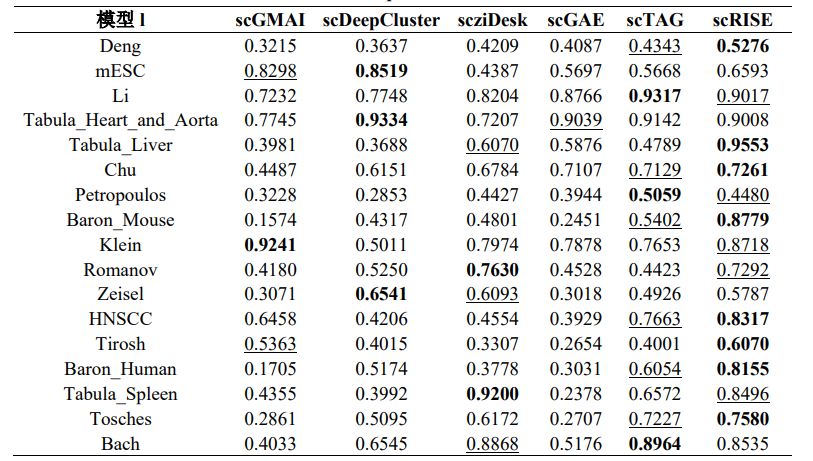
注:每个数据集的最高 ARI 值以粗体表示,第二高的 ARI 值以下划线表示。
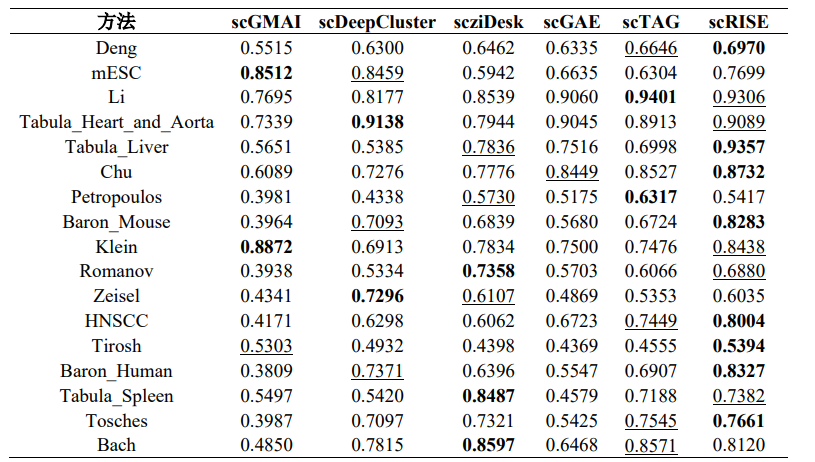
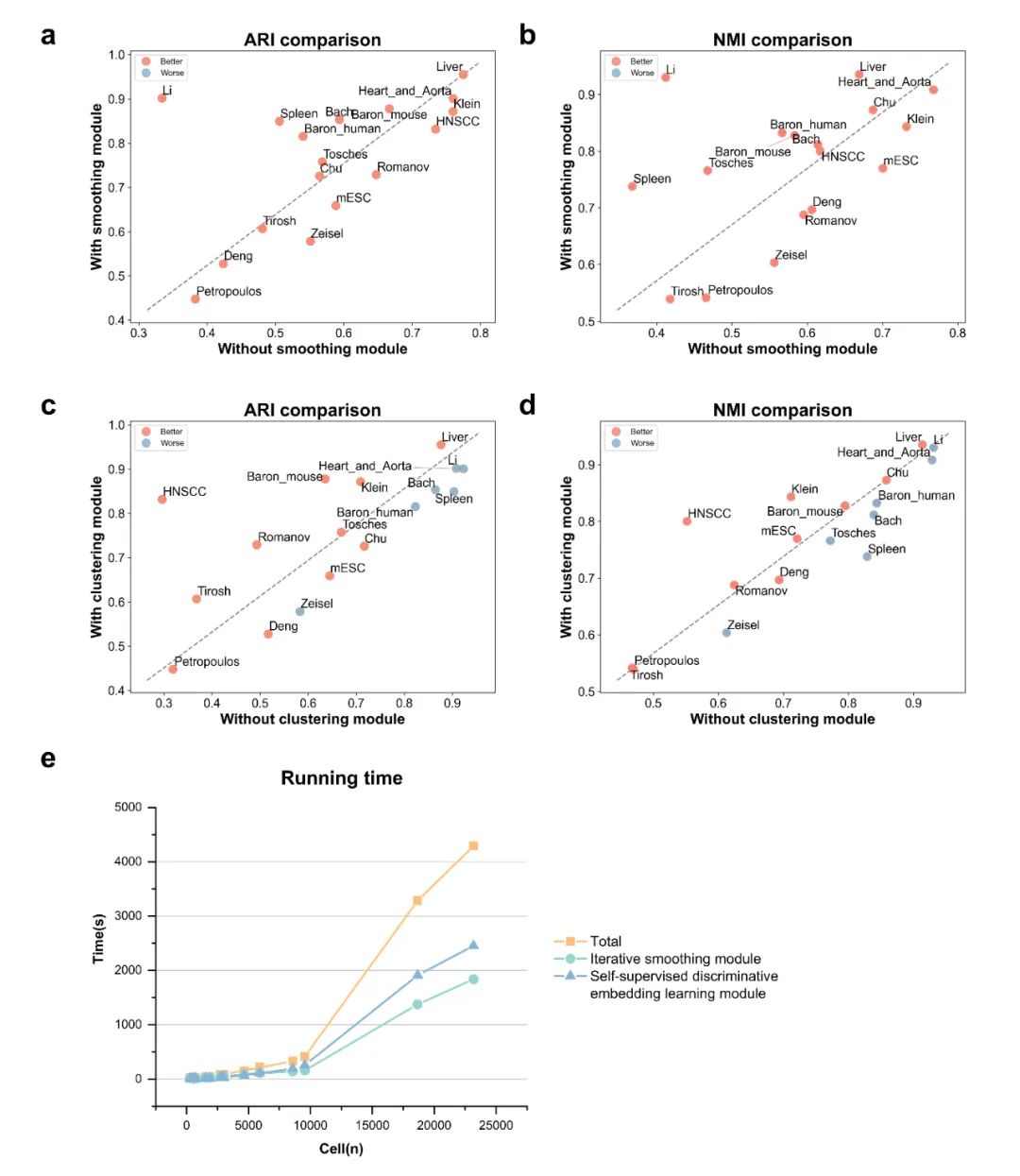
2. 前瞻性研究:头颈鳞状细胞癌中的信息基因挖掘
信息基因是一类在不同细胞类型中表达差异显著的基因集,它们可以用于区分不同的细胞类型。在单细胞RNA测序(scRNA-seq)分析中,通过分析基因表达数据可以鉴定信息基因,并用于确定不同的细胞类型。本研究中,将NMF分解得到的细胞特征矩阵替换为scRISE模型得到的细胞簇特征矩阵,并重构原始的scRNA-seq数据,得到一个包含scRISE模型提取的簇特征的新细胞-基因表达矩阵。然后,使用Lasso回归进一步分析这个新的细胞-基因表达矩阵,以鉴定能够在不同细胞亚型之间做出最佳区分的信息基因。
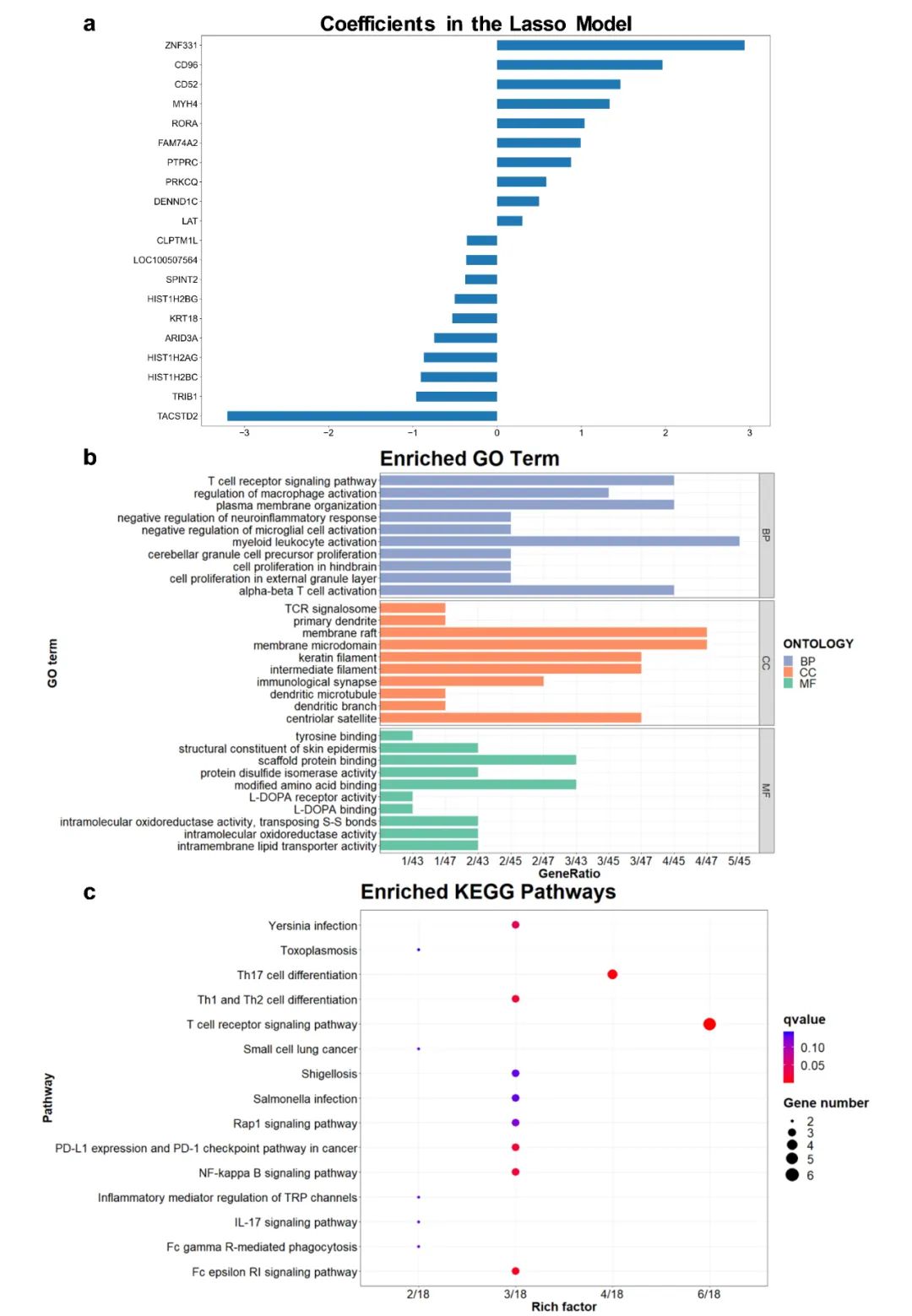
在头颈鳞状细胞癌 (HNSCC) 数据集中,通过设置正则化强度 (λ) 为0.001的Lasso回归提取信息基因,得到62个信息基因。接下来,对获得的62个信息基因进行进一步的基因本体 (GO) 和KEGG富集分析(图3),以探索它们的功能特性,寻找富集的生物过程,并揭示潜在的生物途径。
图3b展示了基因在GO富集中的分布情况,根据生物过程、细胞组分和分子功能三个类别的p值,列出了前10个最重要的GO术语。在生物过程类别中,最常见和富集的GO术语是‘髓系白细胞激活 (Myeloid Leukocyte Activation) ’。在细胞组分类别中,最富集和集中的GO术语是‘膜筏 (Membrane Raft) ’和‘膜微域 (Membrane Microdomain) ’,它们都是富含胆固醇和鞘磷脂的质膜特殊区域。在分子功能类别中,最富集和集中的GO术语是‘支架蛋白结合 (Scaffold Protein Binding) ’。这些结果提供了62个信息基因功能特性的概览,并揭示了它们之间的相互关系,揭示了潜在的生物过程1-3。
图3c展示了KEGG富集的相关途径,按调整后的p值排序,显示了前15个途径。‘T细胞受体信号通路’涉及与T细胞受体激活和下游信号传导相关的一系列蛋白质和分子。在肿瘤免疫逃逸中,‘PD-L1表达和PD-1检查点途径’是一个重要途径。在癌症治疗中,通过抑制PD-L1和PD-1途径增强T细胞免疫活性已成为重要的治疗策略4-6。
这些结果表明,scRISE能够捕捉scRNA-seq数据的关键表示和模式,是一个在生物学研究中解释生物过程的有效分析工具。
本研究提出了一种针对单细胞RNA测序 (scRNA-seq) 数据的深度学习聚类方法,名为scRISE。该方法采用了拉普拉斯数据平滑和自适应学习技术,具有多方面的创新点:首先,该方法使用自编码器来学习并重建数据,无需预设数据分布;其次,引入拉普拉斯平滑滤波器有效降低噪声,提升数据质量而不损失维度;再次,通过迭代循环增强单细胞数据的准确性和稳定性,提高聚类准确性;最后,自适应编码器通过构建相似性矩阵和选择样本对,提取数据的低维特征,增强聚类效果。此外,scRISE在HNSCC数据集上的应用展示了其在临床研究中的潜力和实用性。
综上所述,scRISE模型在单细胞RNA测序数据聚类方面展现出优异的性能,它不仅能够提供精确且具有生物学意义的聚类结果,还表现出了良好的泛化能力和可扩展性。scRISE模型作为一个功能强大的工具,将极大地促进单细胞生物学研究的深入发展,为探索细胞异质性和细胞状态的动态变化提供新的视角和方法。
谢金欣,阮珊珊为本研究的共同第一作者,参与工作的还有屠铭燕,袁珍,胡建国同学,通讯作者为李诗良教授和李洪林教授。该工作得到了中国国家自然科学基金 (82425104,82150208,82173690) 和国家重点研发计划 (2022YFC3400501, 2022YFC3400504) 的资助。

